RUSHING TO JUDGMENT: FUNCTIONALITY IN NONCODING OR “JUNK” DNA
by
Timothy G. Standish Geoscience Research Institute
WHAT THIS ARTICLE IS ABOUT
The 1960s discovery that much nuclear DNA in eukaryotic cells does not code for proteins was quickly interpreted as evidence for the evolution of eukaryotic genomes. Papers were published suggesting a nomenclature reflecting evolutionary assumptions about this “junk DNA.” Noncoding DNA was also used as evidence for the selfish gene theory popularized by Richard Dawkins and others. As many important functions played by noncoding DNA have come to light, the assumption can no longer be made that it represents DNA potsherds of evolution. Now the assumption of functionality in what was once called junk DNA is widespread, but its interpretation within a Darwinian framework remains. Thus, what was once touted as evidence of life’s evolutionary history because of its lack of function is now interpreted as evidence of the same thing because it is functional. This experience calls into question how much data actually unambiguously support Darwinian evolution, what evolutionary theory actually predicts, and how data can be used to check its predictive power.
INTRODUCTION
During the late 1960s papers began appearing that showed eukaryotic DNA contained large quantities of repetitive DNA which did not appear to code for proteins (i.e., Britten and Kohne 1968). By the early 1970s, the term “junk DNA” had been coined to refer to this noncoding DNA (i.e., Ohno 1972). Junk DNA seemed like an appropriate term for DNA cluttering up the genome while contributing in no way to the protein coding function of DNA; yet there seemed to be so much of this noncoding DNA that its significance could not be ignored. One measure of the importance attributed to these noncoding sequences was the awarding of the 1993 Nobel Prize in Medicine and Physiology to Richard Roberts and Phillip Sharp recognizing their 1977 discovery of introns (Chow et al. 1977, and Berget, Moore and Sharp 1977). These DNA sequences interrupt coding sequences and do not code for proteins themselves. In recent lists introns have been categorized as junk DNA along with other noncoding DNA (i.e., Nowak 1994).
Two lines of evidence pointed toward noncoding DNA’s lack of functionality: first, significant variation in noncoding DNA is evident between closely related species and even within species (i.e., Zeyl, Bell and Green 1996). This variation is so great that it is used to produce DNA fingerprints that can differentiate between individual humans and individuals in many other species (Moxon and Wills 1999, Higgins 1999, Baker et al. 1993, Turner et al. 1992, Smith et al. 1990, Jeffreys 1988, Jeffreys, Wilson and Thein 1985). Conservation of protein (and thus DNA) sequences is a hallmark of coding functionality (Lewin 2000); consequently the presence of variability is assumed to mean a given stretch of DNA is noncoding. Initially it was assumed that if DNA did not code for protein, or certain specific RNAs, it was nonfunctional. The second line of evidence was somewhat more direct: mutation of some noncoding DNA did not produce significant changes in phenotype (Nei 1987 discusses this, but also points out that there are some constraints on evolution of noncoding regions). If the DNA did anything, then changing it should change the organism in some way. Some small change in DNA sequences may have little impact, but major and extensive mutation could reasonably be expected to impact any coding function. In the case of some “junk” DNA sequences, seemingly major changes produced no apparent impact.
This paper will document changes in the perceived meaning and role of noncoding DNA, starting with the initial view that an organism’s genome could be viewed in much the same way as archaeologists view middens containing refuse from the past. In this view, noncoding DNA represents the broken remains of old genes that no longer function, mixed in with dust-like repetitive sequences that have blown in and multiplied. Thus, noncoding DNA, as long as it lacks function, may be mined for evidence of life’s distant past and support for the argument that a Designer was not involved in creation.
It is important to emphasize that this logic hinges on lack of knowledge concerning function of noncoding DNA. Functional sequences come under the influence of selection which biases the data to such an extent that a clear interpretation of past history is impossible. Selection may act like grave robbers who remove all precious metals and stones, biasing the record to appear that a culture lacked access to these resources. A second assumption deals with the nature of the Designer, who, if He exists, cannot have cluttered up the genomes of organisms with superfluous sequences. Finally, the mechanism of evolution — the “Blind Watchmaker”, as Richard Dawkins calls it (1986) — could, and in fact would, produce cobbled-together genomes full of bits and pieces, some functional, and many simple clutter. This paper will demonstrate that, as data have accumulated, clear roles for non-coding DNA once thought to lack function have been found, and show that noncoding DNA is as consistent with design theory, perhaps more so, than it ever was with Darwinism.
WHAT IS JUNK DNA?
Because of confusion about the definition of junk DNA, the topic is difficult to discuss unless we first develop a working definition. In the most general use of the term, “junk DNA” is noncoding DNA — DNA that does not directly code for a protein product or specific RNA products like tRNA and rRNA. This noncoding DNA was initially assumed to be functionless, and thus noncoding DNA will be used as the definition of junk DNA in this paper. The meaning of “junk DNA” has become restricted significantly in recent years as the functionality of much of what was once considered junk has become obvious. Most modern genetics and biochemistry texts avoid the term. Even when junk DNA is mentioned, it is frequently given significantly different definitions. For example, Lodish et al. (1995, p 307) called it “extra DNA” for which no function has been found, and then footnotes the comment, “we do not use this term.” Two dictionaries of biological terms (Stenesh 1989, and King and Stansfield 1990) call it “selfish DNA.” In the early 1990s the term “selfish DNA,” coined in the early 1980s (Orgel and Crick 1980, Orgel, Crick and Sapienza 1980), was popularized by Richard Dawkins (1989, p 366) in his book The Selfish Gene.
TYPES OF NONCODING DNA
At least nine classes of DNA were once thought to be functionless. Each of these has been referred to at one time or another as junk, and all were included in a list of types of junk DNA compiled by Nowak
(1994). These nine types can be grouped into three larger groups: 1) untranslated parts of RNA transcripts, 2) repetitive DNA sequences, and 3) other noncoding sequences.
1. Untranslated Parts of RNA Transcripts
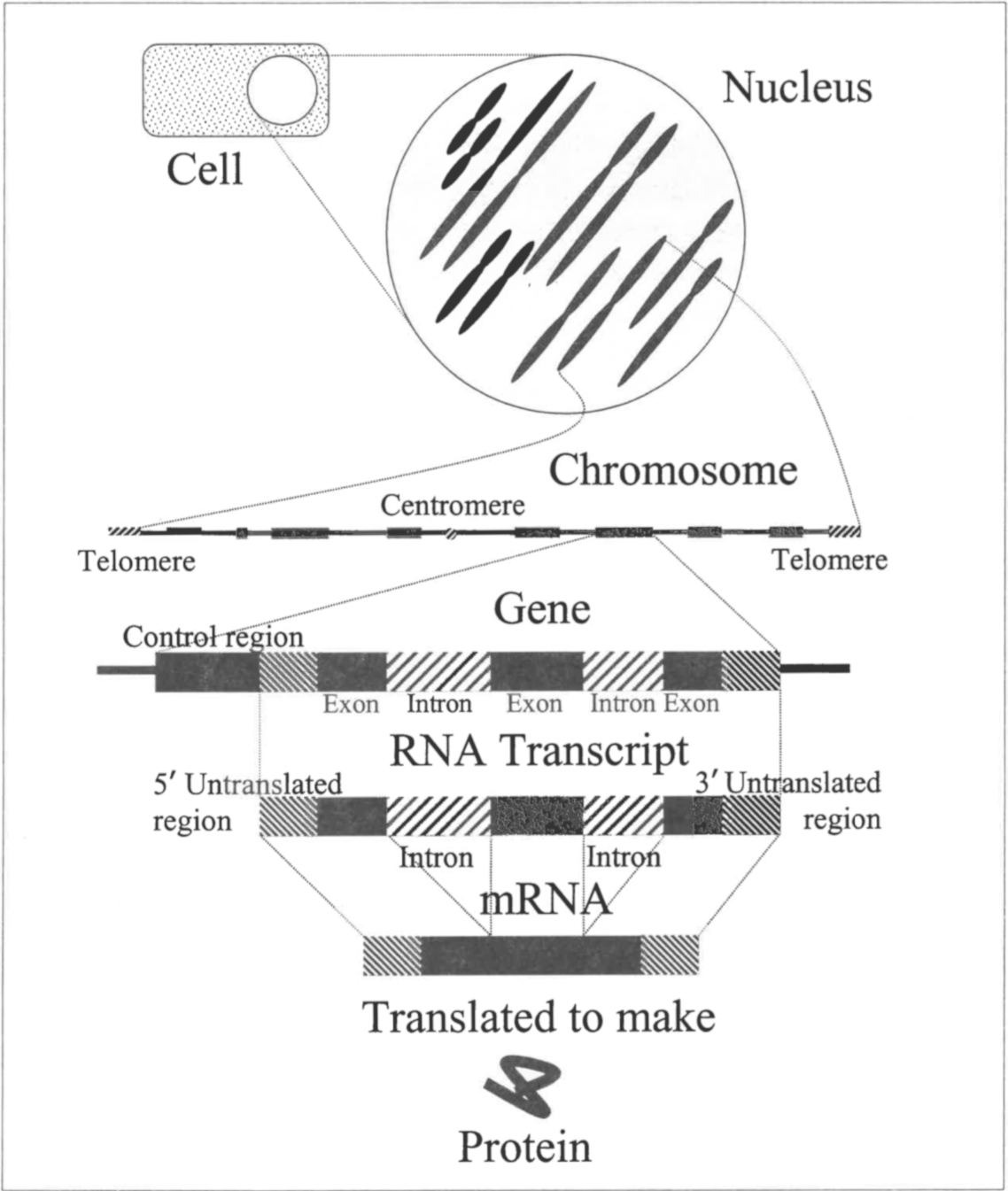
Not all RNA transcribed from DNA actually codes for protein (see Figure 1).
Figure 1. Only a small portion of DNA codes for proteins. Chromosomes, which are found in the cell nucleus, contain long linear stretches of DNA. Within the DNA are genes, but stretches of DNA between the genes do not code for proteins. At the beginning of genes (and frequently elsewhere) are control regions which play a role in regulating gene expression. Following the control region is the part of the gene which is transcribed to RNA. The RNA transcript starts with a 5' untranslated region followed by exons and introns and ends with a 3' untranslated region. Introns are removed to make the exons contiguous, and these contiguous sets of exons code for eukaryotic proteins. The protein is made by ribosomes which translate the RNA message contained in exons.
Initial eukaryotic RNA transcripts produced by RNA polymerase II are called heterogeneous nuclear RNA (hnRNA). Before hnRNA can be exported from the nucleus as mRNA, it must first be processed to remove introns and make other modifications. Parts of the hnRNA that are removed do not code for the protein being produced, but even parts of the mature mRNA do not code for protein. Three noncoding parts of hnRNA are never translated: 1) introns (removed during RNA processing); 2) the 5' untranslated region; and 3) the 3' untranslated region. The latter two leave the nucleus as part of the mRNA. It is only the coding portion of mRNA, referred to as exons because they exit the nucleus, that carry genetic information defining a protein’s amino-acid sequence. This code is translated into a protein in the cytoplasm of eukayrotic cells. Introns that are removed from hnRNA were thought to be junk cluttering the transcript which must be cast aside before the useful coding part of RNA transcripts can be translated.
Nucleic acids are always read in a specific direction, starting at the 5' end, and proceeding toward the 3' end. The 5' and 3' untranslated regions lie at each end of mRNA. Ribosomes, the organelles that translate the coding portion of mRNA into protein, attach first to the 5' end and slide along mRNA in the 3' direction until they reach a start codon signaling the beginning of a protein. Translation from mRNA to protein by ribosomes continues in the 3' direction from the start codon to the first stop codon. It seems reasonable to assume that the mRNA 5' end must play an important role in providing a ribosome attachment site and, this has been demonstrated (Lewin and Siliciano 1997). A function is not as immediately obvious for the mRNA 3' end which follows the stop codon signifying the end of the protein coding region. These 3' untranslated regions, because of their apparent lack of obvious function, have been classified as junk DNA.
2. Repetitive DNA
A surprisingly large proportion of eukaryotic DNA is made up of short sequences repeated many times. These repeated sequences seem too short to code for proteins and are not known to be transcribed. There are five commonly recognized major classes of repetitive DNA:
1) Satellites, also called simple-sequence DNA. These are made up of many (up to 105) tandem repeats of a short DNA sequence, and seem to be concentrated in heterochromatin at the ends (telomeres) and centers (centromeres) of chromosomes. There are at least 10 types of human satellite DNA. Typically they make up 10-15 % of mammals’ genomes.
2) Minisatellites are similar to satellites, but are scattered throughout the genome in clusters of fewer repeats.
3) Microsatellites are shorter still than minisatellites.
4) Short Interspersed Elements (SINEs), like miniand microsatellites, are found distributed throughout the genome, but differ in being single units of DNA about 300 bp (base pairs) in length, instead of repeated shorter units. An example is the human Alu SINE which occurs in the range of 300,000 times (Lewin 2000) making up about 5 % of the human genome (Deininger 1989). One of the interesting properties of SINEs is that they appear to move about in the genome.
5) Long Interspersed Elements (LINEs) are longer than SINEs, up to 7,000 bp — but typically about 6,500 bp — and, like SINEs, may move about in the genome. In mammal genomes there are 20,000-50,000 copies of L1, the most common LINE family (Lewin 2000).
3. Other Noncoding Sequences
Pseudogenes resemble genes, but are not known to be translated into functional proteins. Two classes of pseudogenes have been identified. The first class, unprocessed pseudogenes, resemble normal eukaryotic genes in all respects, but appear to have mutated and become functionless. Processed pseudogenes constitute a second class of pseudogenes. These unexpressed sequences resemble known genes with the introns removed. They appear to have been first transcribed as hnRNA from a functional gene, then processed into mRNA with the introns cut out and then reverse transcribed to make a DNA copy which was then inserted into the organism’s genome at locations independent of the original gene. Both classes of pseudogenes typically contain stop codons within all possible reading frames; thus only fragments of the protein they seem to code for would be produced if they were expressed.
It is not known if pseudogenes are expressed in any way, and because of their assumed history, Lewin (2000) refers to them as “dead ends of evolution.” Interestingly, there are no obvious explanations for why certain gene families have pseudogenes while others do not. More commonly expressed genes may be more likely to have more pseudogenes. The problem with this explanation is that to be inherited, the events necessary to produce a processed pseudogene must occur in the germ cells. It seems unlikely that genes would be expressed in germ cells, which are not known to actively transcribe genes for which processed pseudogenes have been identified. In one exceptional case, a mouse ribosomal protein gene has approximately 15 related processed pseudogenes.
A diverse set of noncoding DNA is represented by heterogeneous nuclear RNA, a mixture of RNAs of varying lengths found in the nucleus. According to Nowak (1994) approximately 25 % of the hnRNA is premRNA that is being processed; the source and role of the remainder is unclear.
PROBLEMS WITH JUNK DNA
Noncoding DNA makes up a significant portion of the total genomic DNA in many eukaryotes. For example, older sources estimate 97 % of the human genome to be noncoding DNA (Yam 1995), while the recently published sequence data increases the estimates to 98.9% noncoding DNA (Venter et al. 2001). These estimates present problems for both intelligent design and naturalistic/evolutionary models of the history of life.
THE PROBLEM FOR DESIGN THEORY
It is difficult to imagine a Designer creating organisms exhibiting elegant efficiency at the gross level, but scattering superfluous molecular debris throughout DNA coding for higher levels of organization. Such inconsistency contradicts the argument that organisms are complex and efficient to such a degree that intelligent design, rather than random events coupled with natural selection, best explains their origin. If design predicts efficiency and noncoding DNA is nonfunctional, then noncoding DNA must be evidence against genome design and for a more haphazard mechanism of origin.
Prominent evolutionists have eagerly proclaimed noncoding DNA to be molecular debris left over by the process of evolution. As mentioned previously, Dawkins (1989) and, much earlier, Orgel and others (1980; and Orgel, Crick and Sapienza 1980) proposed that evolution does not occur at the phenotypic level, but at the molecular level. Successful genes are “selfish” in that they “care” only about perpetuation of their own sequence. In perpetuating themselves, “genes” that do not compromise their “host’s” fitness will proliferate relative to those that decrease host fitness. In this scheme some genes behave in a parasitic manner, perpetuating themselves while not significantly impacting host fitness. This view deconstructs organisms to the point that they are merely conduits for the preservation and proliferation of some segments of DNA, which runs directly contrary to the belief that the constituent parts of organisms all work together to enhance fitness of the whole. Highly repetitive and repetitive DNA sequences, including LINEs, SINEs, and the various satellite DNAs, are assumed to represent these functionless “selfish genes” that exist only for self-perpetuation.
Brosius and Gould (1992) moved boldly during the early 1990s to define the terms used for noncoding DNA in such a way that the data are first interpreted as evidences of evolution and then named. If their terminology had been adopted, any interpretation of the data outside the Darwinian paradigm would first have required a redefinition of the terminology used in discussing the data. They stated: “We wish to propose a general terminology that might aid the integrated study of evolution and molecular biology.” Their proposed system of nomenclature assumed that noncoding DNA represents what was once functional may be functional again, but is currently functionless.
This “genomenclature” was challenged and even greeted with some ridicule at the time of publication. Graur (1993), in what must be one of the most amusing letters ever published in Nature, called genomenclature: “A cruel joke at the expense of the vocal chords of molecular biologists and the integrity of the English language.” Brosius and Gould used terms like “nuon,” meaning any definable stretch of nucleic acids, and “protonuon,” meaning a stretch of nucleic acids with the potential to be recruited as a new gene via mutation and selection. Because these terms (and others) sound suspiciously like the physics terms “muon” and “proton,” Grauer went on to accuse Brosius and Gould of “a clear fit of physics envy.” Grauer’s main objection, however, was to the tonguetwisting nature of genomenclature, not the underlying assumption that noncoding DNA represents debris from the evolutionary past (this is pointed out in Brosius and Gould 1993).
Despite the ridicule, Gould continued to see noncoding DNA as both debris left over during the process of evolution and raw material for future evolution. In a New York Times opinion piece on the Human Genome Project, he stated:
Our 30,000 genes make up only 1 percent or so of our total genome. The rest — including bacterial immigrants and other pieces that can replicate and move — originate more as accidents of history than as predictable necessities of physical laws. Moreover, these noncoding regions, disrespectfully called ‘junk DNA,’ also build a pool of potential for future use that, more than any other factor, may establish any lineage’s capacity for further evolutionary increase in complexity (Gould 2001).
Walter Gilbert and others (Gilbert and Glynias 1993; Dorit and Gilbert 1991; Dorit, Schoenbach and Gilbert 1990) have promoted the idea that the exon-intron arrangement in eukaryotic genes represents a means of rapid evolution of functional genes that overcomes the problems represented by the incredible improbability of producing functional proteins via mutation of initially random sequences. In this model, each exon represents a functional domain; and by combining together different, already functional domains, new functional proteins can be made relatively easily. In other words, exons are the prefabricated nuts and bolts that can be used to make any number of functional molecular machines. Introns are functionless DNA that just happen to fall between functional exons. This view appears to be endorsed by Lewin (2000, p 58-62) in Genes VII, among the most respected molecular biology texts available.
THE PROBLEM FOR DARWINISTS
While Darwinists trumpeted noncoding DNA as prima facie evidence against design, they ignored the fact that efficiency is also accepted within the evolution paradigm as a hallmark of organisms. Efficiency is presumed to increase as natural selection eliminates less-efficient members of a population. As inefficiency increases, the burden it imposes is assumed to impact “fitness.” When the impact on fitness becomes biologically significant, selection will eliminate those organisms with systems relatively less efficient than others competing for the same resources. Only efficient organisms can survive in a selective environment.
The large amount of noncoding DNA in eukaryote genomes seems very inefficient. One would think that a trend would be evident in organisms, going from less to more efficient use of DNA. Ironically, the simpler the organism, the greater its efficiency in DNA use, not the opposite (Lewin 2000). The simplest organisms have little or no noncoding DNA. Alternatively, if noncoding DNA provides grist for the evolutionary mill, one might predict that organisms with more noncoding DNA would evolve more rapidly than those with less “extra” DNA as raw material to work with. This has not been demonstrated. Bacteria with relatively compact genomes are known to adapt to environmental changes at startling rates via rapid mutation. It is true that bacteria have very short generation times, and this may contribute to their rapid adaptation. It is also true that some different mechanisms may be in place in bacteria to direct genetic change, but the reality remains that in this diverse group of organisms whose genetic behavior has been extensively studied, biochemical adaptation to changing environments does not seem to require noncoding DNA.
Relative abundance of noncoding DNA can vary significantly between closely related organisms (see Martin and Gordon 1995, and Sessions and Larson 1987 for examples of this), indicating that changes in the amount of noncoding DNA is an easy evolutionary step. If it is easy to change the quantity of noncoding DNA, the question arises, “Why are those with more than the average amount of noncoding DNA not selected against?” It could be argued that the difference in efficiency between two individuals with varying amounts of noncoding DNA would not be large enough to impact the individual’s reproductive success, but this is a troubling argument that is unsupported by the data.
Making and maintaining DNA requires significant energy input on the part of cells. Not only does the cell have to provide the deoxynucleotide building blocks for extra unneeded DNA, but also enzymes to polymerize and proofread newly made DNA, gyrases to unwind the template DNA, DNA repair enzymes, and so on. Factor all that into the 75 trillion cells in an average human with six billion bases in each nucleus, and the cost becomes potentially significant, even though the cost of other cellular activities may have a much greater direct cost in terms of energy.
The problem of wasted energy would be so much greater if some “junk” DNA were translated, an apparent requirement if it is to serve as a resource for evolution of novel new proteins. Akashi and Gojobori (2002) discuss the cost of polypeptide production and ways in which proteins, particularly those most commonly expressed, are optimized to utilize amino acids with the lowest metabolic cost possible. Clearly, if selection is sensitive enough to adjust specific amino acids within proteins to lower the energy cost of their production, then it should be sensitive enough to eliminate production of any “junk” proteins. It also follows that any DNA sequences that do not provide a selective advantage, especially if they constitute a significant majority of an organism’s genome, should represent a significant metabolic cost and thus be selected against.
It cannot be argued that genome size has no phenotypic impact. Sessions and Larson (1987) have shown that, at least in some closely related salamander species, genome size is negatively correlated with the rate of development. Martin and Gordon (1995) suggest that the large amount of DNA in the nucleus of obligate neotenic salamanders slows development, increases cell size and slows metabolism which they suggest improves survival in cold-water environments. Supporting the theory that increased genome size slows development, Jockusch (1997) showed that genome size is positively correlated with embryonic development time.
Another example of phenotypic change correlated with variation in nuclear DNA size is evident in populations of the flowering plant Silene latifolia. In this plant, genome size shows a significant negative correlation with calyx diameter, a trait of clear ecological importance (Meagher and Costich 1996). Vinogradov (1997) has shown that resting metabolic rate in passerine birds is negatively correlated with increased nuclear DNA when body size is held constant. It is noteworthy that these papers emphasize the supposed evolutionary significance of noncoding DNA, and contradict the assumption that it lacks function. This at least partially disqualifies the previous argument that lack of function in noncoding DNA supports the idea that it is molecular debris of the evolutionary process. Whatever the source, much DNA appears to have a significant phenotypic impact upon which selection may act, whether or not it directly codes for proteins or controls their expression.
Having unneeded DNA presents a potential danger to cells. It is not inconceivable that mutations could occur, resulting in production of noncoding RNA, some of which may interfere with production of essential — or at least beneficial — RNAs and, if they code for them, proteins. If “junk proteins” were made, their production would, at best, waste a cell’s resources or, at worst, alter the activity of other proteins. Darwinists suggest that production of new proteins from old noncoding DNA is the very mechanism by which some new genes were produced. This postulated production of “junk” proteins via genes whose expression is not tightly controlled presents a potential danger to cells both by sapping the resources of the cell for a non-productive task and also because the protein may have functions that interfere with the normal function of other essential components of the cell. Nyolase produced by Flavobacterium has been presented as an example of a new functional protein arising from a sequence (in this case assumed to be a formerly unread reading frame) which did not previously code for any protein (Ohno 1984). If functional proteins can spring forth from previously noncoding sequences, they need not all be adaptive; in fact, harm to the cell appears a far more likely outcome.
Loss of functionless DNA would seem to be a relatively easy evolutionary step. Gaining DNA may be more difficult, although data exist which are consistent with the theory that increases in the number of copies of some DNA stretches has occurred as a result of imperfect crossing over during meiosis prophase I. Alternative explanations of these repeats may be equally consistent with the data, but the important point for this argument is that DNA which is not a normal part of an organism’s genome has been shown to be rapidly lost. For example, Petrov and Hartl (1998) have shown that, at least in Drosophila species, functionless DNA disappears after only a few generations. This appears to be analogous to the vision loss observed in some fish and other organisms that live in caves, or the loss of flying ability observed in birds that live on isolated islands. The conventional explanation is that without selective pressure to maintain them, these abilities are lost. In caves where there is no light, sight provides no selective advantage. Similarly, flight provides little advantage in the absence of predators and presence of abundant marine food around islands. Apparently, at least in Drosophila, extra DNA, like sight and flight, will not be maintained in the absence of selective pressure to maintain it.
The fact that DNA not normally part of a specific genome is easy to lose, combined with evidence that increases in genome size significantly impact phenotype, calls into question the idea that noncoding DNA does not impact fitness enough for natural selection to work on it. These data, combined with the logical inference that noncoding DNA may produce RNA or protein products that negatively impact fitness, all call into question the idea that noncoding DNA represents a currently functionless record of the phylogenetic history of organisms which has been passed down over many generations.
For both intelligent design theorists and Darwinists, noncoding DNA presents a problem if it is really functionless. Intelligent design assumes that a wise Designer would not add functionless rubbish to His creation. Evolutionists assume some function, exemplified in Brosius and Gould’s (1992) nomenclature, if not in the present, at least as remnants of past functionality and raw material for the future. Assuming that noncoding DNA lacks function appears to violate the basic scientific assumption that what is seen in nature exhibits some purpose which can be determined through observation and experimentation. Enthusiasm for absence of function in noncoding DNA appears to have sprung more from philosophical presuppositions, than a careful analysis of data and their implications for Darwinism. If any functionality was to be assigned to noncoding DNA, it was to be done within the context of its role in evolution, not on the basis of any immediate benefit to the organism bearing it in its nucleus.
EVIDENCE OF FUNCTIONALITY IN NONCODING DNA
Both direct and indirect evidence show that functionality is present in some noncoding DNA. One way to look for potential function is to see if DNA sequences exhibit characteristics of other sequences known to be functional. Using this approach, sequences known to code for proteins and those that do not have both been shown to exhibit characteristics of an information carrying code. Searls (1992, 1997) suggested that DNA exhibits all the characteristics of a language, including a grammar. As early as 1981 (Shulman, Steinberg and Westmoreland.1981) and in later papers (i.e., Michel 1986), statistical methods were published for obtaining coding sequences out of the morass of noncoding DNA. More recently statistical studies utilizing neural networks have been used to locate protein coding regions (Uberbacher and Mural 1991, Granjeon and Tarroux 1995). This work concentrates on finding statistical patterns to distinguish coding from noncoding sequences; they do not show that noncoding sequences still contain information, only that they exhibit a statistical signature. More direct work has been reviewed by Yam (1995). Mantegna et al. (1994 and 1995; also see Flam 1994, Havlin et al. 1995, and Peng e al 1995) applied a method for studying languages (Zipf approach) to the study of DNA sequences and suggested “noncoding regions of DNA may carry biological information.” While this paper has not gone unchallenged (see Tsonis, Elsner and Tsonis 1997; Konopka and Martindale 1995; Yam 1995; Chatzidimitriou-Dreismann, Streffer and Larhammar 1996), it does suggest that DNA should be examined for functions other than protein coding.
Aside from protein coding, DNA sequences may include signals controlling replication and other aspects of the cell lifecycle. Manuelidis (1990) suggests that during interphase (ordinary cell activity) chromosomes are localized in specific parts of the nucleus in different cell lines due to three-dimensional structure imparted to them by folding of “junk” DNA. This three-dimensional structure may also “index different genetic compartments for orderly transcription and replication.” More recent work by Macera et al. (1995) has shown that noncoding DNA may play a role in the suppression of genes and suggests that some clinical conditions result from changes in noncoding DNA. Reinhart et al. (2000) have shown that a short RNA sequence regulates developmental timing in Caenorhabditis elegans. Eyre-Walker (1999) has shown evidence for selection on noncoding DNA that varies its GC content. Earlier work by Martin et al. (1984) discussed a mouse interspersed repeat that, “...evolves as if it encodes a protein.” This seems to imply some level of functionality. If selection is operating on a noncoding DNA region, this region must have some impact on fitness. Related to this thought is research reported by Koop and Hood (1994) showing surprising sequence homology between long regions of corresponding mouse and human noncoding DNA, again implying function and selection to maintain the sequence.
After the excitement about noncoding DNA in the early 1970s, many special examples of functional noncoding sequences have been found. Every untranslated part of hnRNA and mRNA has been found to have a function in at least some transcripts. Some introns contain other genes that are expressed independently of the exons they separate. Thus, as long as the coding strands for both genes are the same, they will always be transcribed together. In addition, the work of Thomas Cech (Cech 1985, Kruger et al. 1982, Zaug, Grabowski and Cech 1983) has shown that introns are not noncoding stretches of RNA transcribed from equally functionless DNA but, in some cases, act in complex ways resembling protein enzymes as they splice themselves out of pre-mRNA. These segments of DNA, once thought of as merely interrupting the important parts of eukaryotic genes, are now found to play an active role in removing themselves from the gene transcript. This does not show a coding role for the introns, but reveals a level of complexity and potential functionality previously unanticipated. Not all introns have been shown to contain these “ribozymes,” but ribozymes should encourage caution before writing off introns as having no function.
It is also worth noting that there is a significant trend toward increased gene size when bacterial genes are compared to those in single-celled eukaryotes. The trend continues when yeast genes are compared with nematode or fly genes, and when these relatively “simple” organisms’ genes are compared with genes from humans and other complex multicelled eukaryotes. While gene size goes up dramatically, only a very small proportion of the increase results from increases in the size of exons which code for protein. The bulk of the increase can be attributed to increase in the number and size of introns (Lewin 2000). This correlation between an increase in introns and an increase in apparent phenotypic complexity needs to be explored further before the role of the introns is assumed to be insignificant.
Specific functions for some introns have been discovered. For example, many introns also code for small nuclear RNAs (snRNAs). These accumulate in the nucleolus, and may play a role in ribosome assembly. Thus the introns that are cut out of the pre-mRNA may play a role in either producing or regulating machinery that translates mRNA’s codons into protein. Zuckerkandl (1997) reviews work showing introns, along with other noncoding DNA, play an important role in repression of genes and the sequential switching of genes during development, suggesting that up to 15 % of “junk DNA” functions in this vital role. A specific example of regulation of expression by an intron sequence involved the suppression of rat osteocalcin gene by the sequence TTTCTTT within the first intron of the osteocalcin gene (Goto et al. 1996). The repressor sequence serves as a negative feedback on expression of the gene.
In artificial settings, RNA has been shown to be capable of repressing the expression of specific genes. This repression has been demonstrated in a heritable manner in the roundworm Caenorhabditis elegans (Grishok, Tabara and Mello 2000). The extent, if any, of RNA inhibition (RNAi) in nature has not been established, but serves as another example of an unanticipated role for RNA which may be related to the kind of negative feedback seen in the TTTCTTT sequence in the osteocalcin intron. A general review of the nature and role of introns when viewed from a design standpoint is given by Bergman (2001).
The obvious role of the 5' untranslated region of mRNA in signaling for ribosome binding has already been mentioned. Untranslated regions at the 3' end of mRNAs have been found to play an important role in the regulation of some gene activity (Wickens and Takayama 1994) and thus clearly engage in an important function. A specific example of function for the 3' untranslated region has been demonstrated in regulation of the human luteinizing hormone/chorionic gonadotropin receptor gene (Lu and Menton 1996). In this case several different mRNA transcripts for the receptor gene are known. The mRNA species with a long 3' untranslated region repress expression of the gene by reducing affinity for ribosomes and reducing the mRNA cytoplasmic half-life. The mRNA species with short 3' untranslated regions increased protein expression, apparently as a result of some other post-transcriptional mechanism of regulation. As new roles played by RNA are discovered and understanding increases of the enzyme-like properties of some RNAs, dismissing hnRNAs which are not precursors of mRNA as lacking in immediate function seems premature. It may be reasonable to predict that as we learn more about the roles of noncoding RNA sequences inside and outside of the nucleus, particularly in control of gene expression, ever-decreasing amounts of it will be consigned to speculative roles in an organism’s evolution.
At the centromere, satellite DNA sequences play a role in assembly of the kinetochore and attachment of spindle fibers during mitosis (Wells 1966). Satellite sequences play an equally dramatic role at chromosome ends where a few nucleotides are lost from telomeres during each replication cycle. Given enough replications, telomeres are eliminated unless the lost nucleotides are replaced. Loss of the telomeres leads to chromo-some shortening, and further replication and shortening results in loss of important functional genes. Damage or loss of these genes may lead to cell senescence. It is speculated that telomere loss is a partial mechanism for aging (see Hodesa 1999 for a review of the relationship between telomeres and aging). In some — but not all — cells, special enzymes called telomerases add satellite DNA to the ends of chromosomes so that there is little or no loss of DNA after replication. Thus, noncoding satellite DNA in telomeres plays an important role in preserving the ends of chromosomes and maintaining functional cell lines. Some suggestions about the role of natural selection working on the length of DNA segments and favoring tandem repeats have been made by Stephan and Cho (1994). In this case, the function of some tandem repeats may be to regulate length, not to code or signal anything else.
Long and Short Interspersed Elements (LINEs and SINEs) appear at first examination to almost perfectly fit Dawkin’s definition of “selfish DNA.” Because of their transposon activity, they seem to pose a hazard to normal gene function. As they move around in the genome they may insert into functional genes disrupting protein coding, or destroying control regions. A number of documented genetic diseases have been shown to be caused by movement of SINEs and LINEs. Individual cases of neurofibromatosis-1 (elephant man disease) are associated with insertion of a SINE, while some instances of hemophilia and Duchenne muscular dystrophy appear to result from disruption of genes by LINES. Aside from destroying genes as they move around in genomes, potential function for at least one SINE has been demonstrated: the Alu SINE has been shown to play a role in control of protein synthesis when cells are stressed (Chu et al. 1998). A role in X chromosome inactivation has also been proposed for the most common LINE, L1 (Lyon 2000). This is based on the observation that, compared to its frequency in autosomes, L1 appears at almost twice the frequency on X chromosomes and is particularly concentrated around the region where chromosome inactivation starts (Bailey et al. 2000). Most recently a potentially very important role in repairing breaks in DNA has been demonstrated for L1 by Morrish et al. (2002).
The absence of a recognized general role for microsatellites may be because this designation is based on sequence characteristics, not function. While the characteristics of different sequences may categorize them as microsatellites, their functions may vary dramatically. Nadir et al. (1996) show evidence that the Alu SINE is associated with A-rich microsatellites and suggest a role for this class of microsatellites in providing targets for Alu insertion. According to this interpretation, Arich microsatellites act as markers for Alu retroposition, thus playing a role in preventing gene disruption by insertion of Alu at inappropriate locations. This may be an important role given the already noted diseases caused by movement of SINEs and LINES.
Clearly, if this interpretation is correct, microsatellites play an important role in the organization of chromatin and, in cooperation with the Alu SINE, may act as part of an elaborate mechanism for the regulation of gene expression. A separate role for microsatellites in organization of chromosomes within the nucleus is suggested by the observation of Gasser and Laemmli (1987) who noted that A and T boxes resembling A-rich microsatellites are found associated with the nuclear scaffold. Attachment of chromosomes to the nuclear scaffold, possibly involving these A and T boxes, is believed to be responsible for arrangement of DNA within the nucleus.
Defects in microsatellites are associated with some types of cancer, although this is assumed to be an indicator of susceptibility to replication errors rather than a cause of cancer (Moxon and Wills 1999). Increase in the number of repeats within microsatellites making up part of the coding portion of some genes has been associated with Huntington’s disease and a number of rare neurological disorders. Variation in the size of triplet repeat microsatellites within genes has been shown to affect gene expression. Interestingly, Moxon and Wills suggest that rather than being the molecular debris of evolution, microsatellites play an active role in the adaptation of bacteria to potentially lethal changes in their environment. Because of the role played by microsatellites in phase variation, Moxin and Wills call bacterial microsatellites “true evolutionary adaptations.” They go on to suggest that microsatellites may play a similar role in the rapid adaptive regulation of eukaryotic genes. This represents a major shift from viewing this class of noncoding DNA as lacking function or as selfish DNA, although it still illustrates the imposition of an evolutionary framework on how data are interpreted.
At least one microsatellite sequence — AGAT — has a demonstrated function in regulation (Weiss and Orkin 1995). This shows that different subclasses of microsatellites may play significantly different, but important, roles (Nadir et al. 1996).
The role of pseudogenes, if there is one, remains problematic. Unprocessed pseudogenes appear to be copies of normal genes which mutated and lost their function over the course of time. Processed pseudogenes appear to be degenerate genes. These present a more problematic picture, particularly in the light of their association with retroposons.
Despite the poor record of the assumption that noncoding DNA is functionless, papers published relatively recently invoke the term “junk DNA” when describing DNA for which no function has yet been determined (see Gardiner 1997 for an example of this). Still, the assumption that noncoding sequences lack function seems to be going out of vogue, and calls are being made to investigate potential functions for even the most unpromising simple repeats (for example see Epplen, Maeueler and Santos 1998). Because the term “junk DNA” is still used to refer to noncoding DNA, much of which is clearly functional, there is some discussion of completely abandoning the term, although no obvious replacement is evident (Kuska 1998a,b), the efforts of Brosius and Gould (1992) having been ignored. However, the term remains in use and a cursory search using PubMed reveals at least 10 instances of its use in titles of papers published in major journals between 1997 and 2001. All of these papers either deal with technical issues associated with noncoding DNA in the general study of DNA sequences, or suggest functions for it. Clearly, while specific functions for all noncoding DNA have not been discovered, the assumption of lack of function no longer dominates the thinking of molecular biologists.
CONCLUSIONS
Much of the excitement surrounding noncoding DNA appears to have been misdirected. In many respects the history of noncoding DNA resembles that of vestigial organs. Evolutionists accepted the assumed lack of function of noncoding DNA as evidence supporting their worldview, even though lack of function is not necessarily a logical deduction from evolutionary theory. Furthermore, an assumption of function does not have to follow from the idea of design. In claiming that noncoding DNA supports evolutionary theory, predictions of functionality reasonably based on that theory had to be ignored.
Darwinists defined what they thought a Designer would do and then presented noncoding DNA as violating that prediction. In doing this three mistakes were made:
1) Terms of the argument were unfairly constrained by defining the Designer in a way that seemed to be contradicted by the evidence. If a Designer exists, He is not compelled to fit any definitions His creatures may want to impose, especially not those definitions that preclude His existence on the basis of what He created. Designers can do whatever pleases them. If this were not so, it would be reasonable to question that automobiles with functionless fins from the 1950s were designed by intelligent beings.
2) A second error involved treating the hypothesis that noncoding DNA lacked function as if it were well-supported by the data, when there were little data. Worse still, the hypothesis was invoked as if it were a fact instead of a tentative interpretation. If noncoding DNA is functional, then the argument that a Designer would not have included functionless junk in the design becomes irrelevant.
3) The final failure was neglecting to examine evolutionary theory to be sure that it does not predict functionality. This failure resulted in a false dichotomy between the predictions made by design versus those made by Darwinism. It might be argued that in this final error some latitude can be given, as evolutionary theory does not always make clear predictions. In fact, it frequently appears to be more robust than other ideas because it can be adjusted to “predict” whatever the data happen to say. As long as noncoding DNA appears functionless, that is what evolutionary theory predicts, but if it is functional, then evolutionary theory provides an equally accommodating framework in which to fit the data.
The history of noncoding DNA serves as a cautionary tale illustrating the danger inherent in ignoring the predictive value of one’s paradigm. Careful evaluation is needed before jumping on a new trend and claiming that it supports one side or the other of the creation-evolution debate. In attempting to discredit creationists, Darwinists ignored the prediction of functionality made by their own theory and the lack of supporting data. Rushing to judgment is never a wise first step when examining the predictions of competing theories in the absence of sufficient data.
LITERATURE CITED
Akashi J, Gojobori T. 2002. Evolution metabolic efficiency and amino acid composition in the proteomes of Escherichia coli and Bacillus subtilis. Proceedings of the National Academy of Sciences (USA) 99:2695-3700.
Bailey JA, Carrel L, Chakravarti A, Eichler EE. 2000. Molecular evidence for a relationship between LINE-1 elements and X chromosome inactivation: The Lyon repeat hypothesis. Proceedings of the National Academy of Sciences (USA) 97:66346639.
Baker CS, Gilbert DA, Weinrich MT, Lambertsen R, Calambokidis J, McArdle B, Chambers GK, O’Brien SJ. 1993. Population characteristics of DNA fingerprints in humpback whales (Megaptera novaeangliae). Journal of Heredity 84:281-290.
Berget SM, Moore C, Sharp PA. 1977. Spliced segments at the 5' terminus of adenovirus 2 late mRNA. Proceedings of the National Academy of Sciences (USA) 74:31713175.
Bergman J. 2001. The functions of introns: from junk DNA to designed DNA. Perspecfives on Science and Christian Faith 53:170-178.
Britten RJ, Kohne DE. 1968. Repeated sequences in DNA. Hundreds of thousands of copies of DNA sequences have been incorporated into the genomes of higher organisms. Science 161:529-540.
Brosius J, Gould SJ. 1992. On ‘genomenclature’: a comprehensive (and respectful) taxonomy for pseudogenes and other ‘junk DNA.’ Proceedings of the National Academy of Sciences (USA) 89:10706-10710.
Cech TR. 1985. Self-splicing RNA: implications for evolution. International Review of Cytology 93:3-22.
Chatzidimitriou-Dreismann CA, Streffer RM, Larhammar D. 1996. Lack of biological significance in the ‘linguistic features’ of noncoding DNA — a quantitative analysis. Nucleic Acids Research 14:1676-1681.
Chow LT, Gelinas RE, Broker TR, Roberts RJ. 1977. An amazing sequence arrangement at the 5' ends of adenovirus 2 messenger RNA. Cell 12:1-8.
Chu WM, Ballard R, Carpick B, Williams BR, Schmid CW. 1998. Potential Alu function: regulation of the activity of double-stranded RNA-activated kinase PKR. Molecular and Cellular Biology 18:58-68.
Dawkins R. 1986. The blind watchmaker. NY: W.W. Norton and Co.
Dawkins R. 1989. The selfish gene. 2nd ed. London: Oxford University Press.
Deininger PL. 1989. SINEs: short, interspersed repeated DNA elements. In: Berg DE, Howe MM, editors. Mobile DNA. Washington DC: American Society of Microbiology, p 619-636.
Dorit RL, Gilbert W. 1991. The limited universe of exons. Current Opinion in Genetics & Development 1:464-469.
Dorit RL, Schoenbach L, Gilbert W. 1990. How big is the universe of exons? Science 250:1377-1382.
Epplen JT, Maueler W, Santos EJ. 1998. On GATAGATA and other “junk” in the barren stretch of genomic desert. Cytogenetics and Cell Genetics 80:75-82.
Eyre-Walker A. 1999. Evidence of selection on silent site base composition in mammals: potential implications for the evolution of isochores and junk DNA. Genetics 152:675-683.
Flam F. 1994. Hints of a language in junk DNA [news]. Science 266:1320.
Gardiner K. 1997. Clonability and gene distribution on human chromosome 21: reflections of junk DNA content? Gene 205:39-46.
Gasser SM, Laemmli UK. 1987. A glimpse at chromosomal order. Trends in Genetics 3:16-22.
Gilbert W, Glynias M. 1993. On the ancient nature of introns. Gene 135:137-144.
Goto K, Heymont JL, Klein-Nulend J, Kronenberg HM, Demay MB. 1996. Identification of an osteoblastic silencer element in the first intron of the rat osteocalcin gene. Biochemistry 35:11005-11011.
Gould SJ. 2001. Humbled by the genome’s mysteries [opinion]. The New York Times, 19 February 2001.
Granjeon E, Tarroux P. 1995. Detection of compositional constraints in nucleic acid sequences using neural networks. Computer Applications in the Biosciences 11:2937.
Grauer D. 1993. Molecular deconstructivism [letter]. Nature 363:490. Grishok A, Tabara H, Mello CC. 2000. Genetic requirements for inheritance of RNAi in C. elegans. Science 287:2494-2497.
Havlin S, Buldyrev SV, Goldberger AL, Mantegna RN, Peng CK, Simons M, Stanley HE. 1995. Statistical and linguistic features of DNA sequences. Fractals 3:269-284.
Higgins M. 1999. Acid test. ABA [American Bar Assn] Journal 85(October):64-67.
Hodesa RJ. 1999. Telomere length, aging, and somatic cell turnover. Journal of Experimental Medicine 190:153-156.
Jeffreys AJ, Wilson V, Thein SL. 1985. Hypervariable ‘minisatellite’ regions in human DNA. Nature 314:67-73.
Jeffreys AJ, Wilson V, Neumann R, Keyte J. 1988. Amplification of human minisatellites by the polymerase chain reaction: towards DNA fingerprinting of single cells. Nucleic Acids Research 16:10953-10971.
Jockusch EL. 1997. An evolutionary correlate of genome size change in plethodontid salamanders. Proceedings of the Royal Society of London Series B 264:597-604.
King RC, Stansfield WD. 1990. A dictionary of genetics. 4th ed. NY: Oxford University Press.
Konopka AK, Martindale C. 1995. Noncoding DNA, Zipf’s law, and language [letter]. Science 268:789.
Koop BF, Hood L. 1994. Striking sequence similarity extending over almost 100 kilobases of human and mouse T-cell receptor DNA. Nature Genetics 7:48-53.
Kruger K, Grabowski PJ, Zaug AJ, Sands J, Gottschling DE, Cech TR. 1982. Self-splicing RNA: autoexcision and autocyclization of the ribosomal RNA intervening sequence of Tetrahymena. Cell 31:147-157.
Kuska B. 1998a. Should scientists scrap the notion of junk DNA? [news]. Journal of the National Cancer Institute 90:1032-1033.
Kusk B. 1998b. Bring in da noise, bring in da junk — the semantics of junk DNA [news]. Journal of the National Cancer Institute 90:1125-27.
Lewin B, Siliciano P. 1997. Genes VI. NY: Oxford University Press.
Lewin B. 2000. Genes VII. NY: Oxford University Press.
Lodish H, Baltimore D, Berk A, Zipursky SL, Matsudaira P, Darnell J. 1995. Molecular cell biology. 3rd ed. NY: W. H. Freeman.
Lu DL, Menon KM. 1996. 3' untranslated region-mediated regulation of luteinizing hormone/human chorionic gonadotropin receptor expression. Biochemistry 35:12347-12353.
Lyon MF. 2000. LINE-1 elements and X chromosme inactivation: a function for “junk” DNA? Proceedings of the National Academy of Sciences (USA) 97:6248-6249.
Macera MJ, Verma RS, Conte RA, Bialer MG, Klein VR. 1995. Mechanisms of the origin of a G-positive band within the secondary constriction region of human chromosome 9. Cytogenetics and Cell Genetics 69:235-239.
Mantegna RN, Buldyrev SV, Goldberger AL, Havlin S, Peng C-K, Simons M, Stanley HE. 1994. Linguistic features of noncoding DNA-sequences. Physical Review Letters 73:3169-3172.
Mantegna RN, Buldyrev SV, Goldberger AL, Havlin S, Peng C-K, Simons M, Stanley HE. 1995. Systematic analysis of coding and noncoding DNA sequences using methods of statistical linguistics. Physical Review E 52:2939-2950.
Manuelidis L. 1990. A view of interphase chromosomes. Science 250:1533-1540.
Martin CC, Gordon R. 1995. Differentiation trees, a junk DNA molecular clock, and the evolution of neoteny in salamanders. Journal of Evolutionary Biology 8:339-354.
Martin SL, Voliva CF, Burton FH, Edgell MH, Hutchison CA (3rd). 1984. A large interspersed repeat found in mouse DNA contains a long open reading frame that evolves as if it encodes a protein. Proceedings of the National Academy of Sciences (USA) 81:2308-2312.
Meagher TR, Costich DE. 1996. Nuclear DNA content and floral evolution in Silene latifolia. Proceedings of the Royal Society of London Series B 263:1455-1460.
Michel CJ. 1986. New statistical approach to discriminate between protein coding and non-coding regions in DNA sequences and its evaluation. Journal of Theoretical Biology 120:223-236.
Morrish TA, Gilbert N, Myers JS, Vincent BJ, Stamato TD, Taccioli GE, Batzer MA, Moran JV. 2002. DNA repair mediated by endonuclease-independent LINE-1 retrotransposition. Nature Genetics 31:159-165.
Moxon ER, Wills C. 1999. DNA microsatellites: agents of evolution? Scientific American 280(1):94-99.
Nadir E, Margalit H, Gallily T, Ben-Sasson SA. 1996. Microsatellite spreading in the human genome: evolutionary mechanisms and structural implications. Proceedings of the National Academy of Sciences (USA) 93:6470-6475.
Nei M. 1987. Molecular evolutionary genetics. NY: Columbia University Press.
Nowak R. 1994. Mining treasures from “Junk DNA” [news; includes related glossary]). Science 263:608-610.
Ohno S. 1972. So much “junk” DNA in our genome. Brookhaven Symposia in Biology 23:366-370.
Ohno S. 1984. Birth of a unique enzyme from an alternative reading frame of the preexisted, internally repetitious coding sequence. Proceedings of the National Academy of Sciences (USA) 81:2421-2425.
Orgel LE, Crick FH. 1980. Selfish DNA: the ultimate parasite. Nature 284:604-607.
Orgel LE, Crick FH, Sapienza C. 1980. Selfish DNA [news]. Nature 288:645-646.
Peng C-K, Buldyrev SV, Goldberger AL, Havlin S, Mantegna RN, Simons M, Stanley HE. 1995. Statistical properties of DNA sequences. Physica A 221:180-192.
Petrov DA, Hartl DL. 1998. High rate of DNA loss in the Drosophila melanogaster and Drosophila virilis species groups. Molecular Biology and Evolution 15:293-302.
Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvie AE, Horvitz HR, Ruvkun G. 2000. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 403:901-906.
Searls DB. 1997. Linguistic approaches to biological sequences. Computer Applications in the Biosciences 13:333-344.
Searls DB. 1992. The linguistics of DNA. American Scientist 80:579-591.
Sessions SK, Larson A. 1987. Developmental correlates of genome size in Plethodontid salamanders and their implications for genome evolution. Evolution 4:1239-1251.
Shulman MJ, Steinberg CM, Westmoreland N. 1981. The coding function of nucleotide sequences can be discerned by statistical analysis. Journal of Theoretical Biology 88:409-420.
Smith JC, Newton CR, Alves A, Anwar R, Jenner D, Markham AF. 1990. Highly polymorphic minisatellite DNA probes. Further evaluation for individual identification and paternity testing. Journal of the Forensic Science Society 30:3-18.
Stenesh J. 1989. Dictionary of biochemistry and molecular biology. 2nd ed. NY: John Wiley & Sons.
Stephan W, Cho S. 1994. Possible role of natural selection in the formation of tandemrepetitive non-coding DNA. Genetics 136:333-341.
Tsonis AA, Elsner JB, Tsonis PA. 1997. Is DNA a language? Journal of Theoretical Biology 184:25-9.
Turner BJ, Elder JF (Jr), Laughlin TF, Davis WP, Taylor DS. 1992. Extreme clonal diversity and divergence in populations of a selfing hermaphroditic fish. Proceedings of the National Academy of Sciences (USA) 89:10643-10647.
Uberbacher EC, Mural RJ. 1991. Locating protein-coding regions in human DNA sequences by a multiple sensor-neural network approach. Proceedings of the National Academy of Sciences (USA) 88:11261-11265.
Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, and 269 other authors. 2001. The sequence of the human genome. Science 291:1304-1351.
Vinogradov AE. 1997. Nucleotypic effect in homeotherms: body-mass independent resting metabolic rate of passerine birds is related to genome size. Evolution 51:220-225.
Wells W. 1996. Don’t write off ‘junk’ DNA. New Scientist 150:19.
Wickens M, Takayama K. 1994. Deviants — or emissaries [news]. Nature 367:17-18.
Yam P. 1995. Talking trash: linguistic patterns show up in junk DNA. Scientific American 272(3):24.
Zaug AJ, Grabowski PJ, Cech TR. 1983. Autocatalytic cyclization of an excised intervening sequence RNA is a cleavage-ligation reaction. Nature 301:578-583.
Zeyl C, Bell G, Green DM. 1996. Sex and the spread of retrotransposon Ty3 in experimental populations of Saccharomyces cerevisiae. Genetics 143:1567-1577.
Zuckerkandl E. 1997. Junk DNA and sectorial gene repression. Gene 205:323-343.